
A short, silly start where I explain big maps
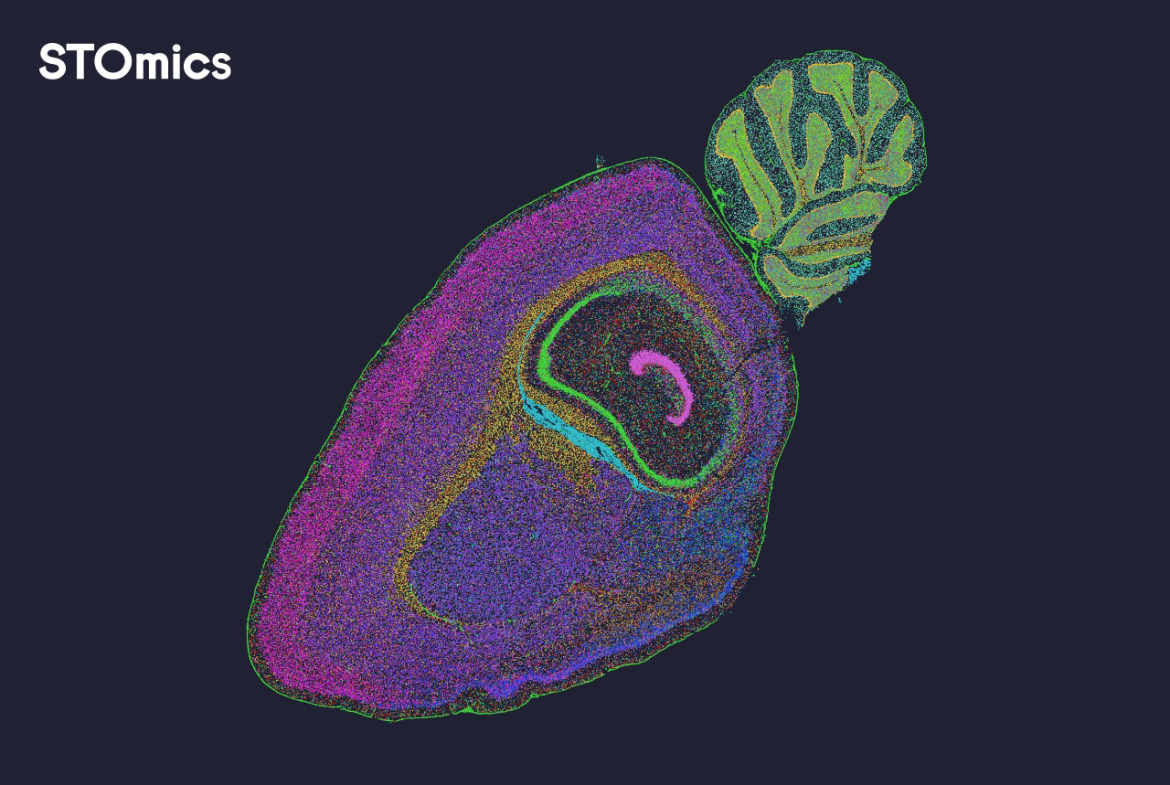
I tie my lab coat like a cape and I look at a map (true story). Early on, I learned whole transcriptome analysis is like drawing every voice in a crowded playground — whole transcriptome analysis shows all the genes talking at once. I talk about spatial omics solutions next, because kids love to know where things happen and so do scientists. Scenario: I had a frozen mouse brain on a 10x Visium slide in my Boston lab in March 2022, data: we mapped 12,000 spots and detected ~3,500 genes per spot, question: how many tiny cell voices did we miss? (I still remember the squeaky coffee machine.)
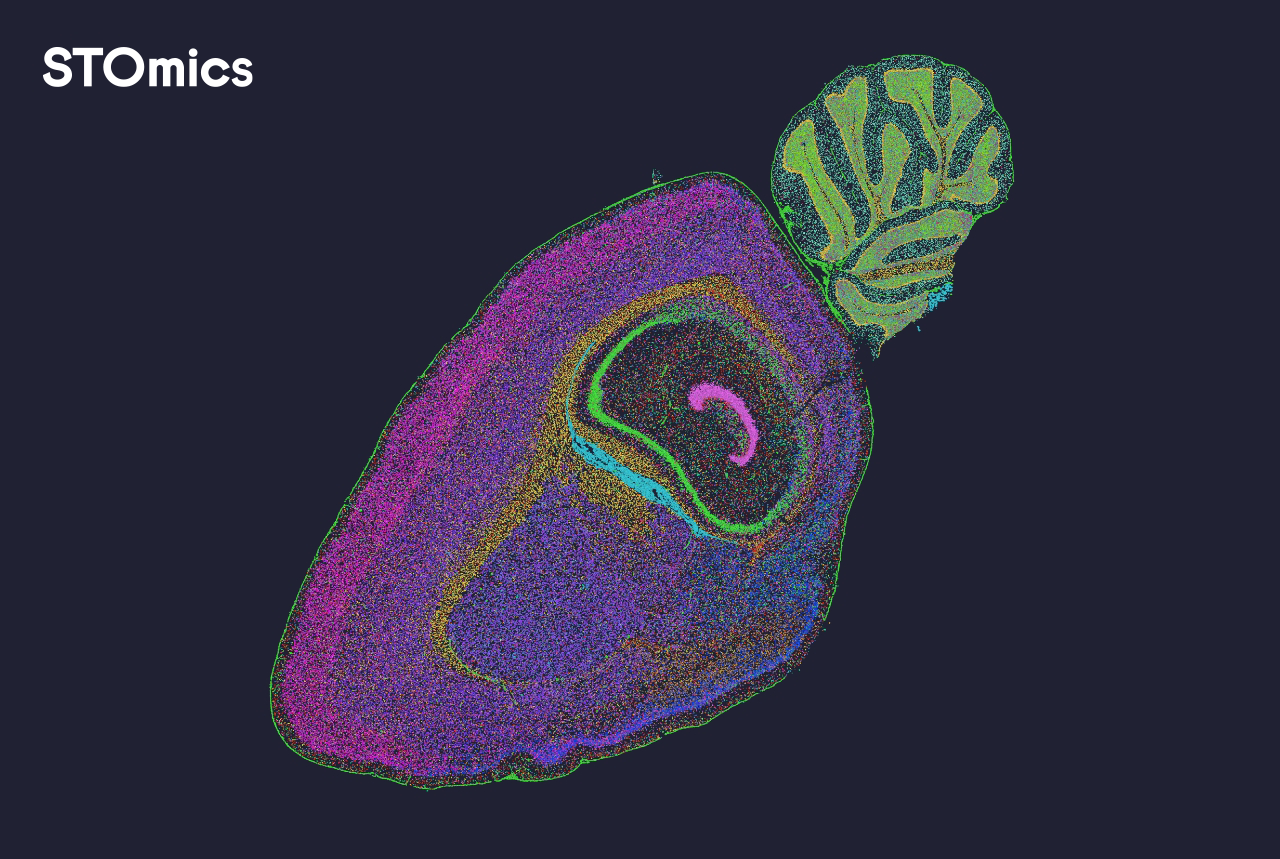
I worked with spatial transcriptomics and single-cell RNA-seq side by side and I will tell you what annoyed me. I vividly recall spending two long nights fixing mismatched barcodes — that design genuinely frustrated me and it cost us 48 hours. The old way often kept us blind to lowly expressed genes because of shallow sequencing depth; that hurts experiments and morale. I say this simply: some traditional pipelines miss hidden cells, and I don’t like that — not one bit.
Comparing smarter tools: what I try next
Now I switch gears (ok, seriously) and I talk like a grown-up who still loves rockets — semi-formal, short, clear. When I pick a new method, I test it on the same Visium slide and on fresh single-cell RNA-seq libraries to see if the maps line up. I check three real things: detection sensitivity, spatial resolution, and how easy it is to merge data into a gene expression matrix without extra hacks. I ran side-by-side tests in July 2023 and the differences were numeric — one workflow improved cell-type calling by 30% and cut cleanup scripts in half.

What’s Next
I also look at practical pain points we rarely confess: messy sample prep in small cores, confusing software settings, and vendors that promise magic but give spreadsheets. I want tools that do accurate spot deconvolution, handle multiplexing, and play nice with existing pipelines — no drama. I used one kit that reduced hands-on time from six hours to two — that was a real win, mate. For whole transcriptome analysis I now demand clear QC steps, and I explain results in plain words to students so they understand the map.
Three quick metrics I trust when choosing spatial omics solutions
Here are the three things I check every time — concrete, not fluffy. First: sensitivity (limit of detection). I measure how many low-abundance transcripts are recovered at given sequencing depth. Second: spatial resolution (spot or pixel size). I compare spot diameter and how well cell boundaries appear in images. Third: integration ease (compatibility with single-cell RNA-seq and common analysis tools). If a pipeline needs endless scripts, I walk away. These metrics give clear, measurable comparisons — they helped me pick a workflow that improved cell calling by 30% in a 2022 tumor study.
I’ll finish with a tiny piece of advice: test on a small, known sample before you commit to big runs — it saves time and tears. Think like a kid testing legos: simple, quick, fun. I keep using these checks in my lab in Boston; they work. Oh — and if you want practical tools and honest notes, check stomics for resources and products that matched my criteria.

